

El Paso, TX Estrés Oxidativo y Defensa Antioxidante
Quiropráctico basado en la ciencia Dr. Alexander Jimenez Echa un vistazo a estrés oxidativo, lo que es, cómo afecta el cuerpo y la defensa antioxidante para remediar la situación.
Esra Birben PhD, 1 Umit Murat Sahiner MD, 1 Cansin Sackesen MD, 1 Serpil Erzurum MD, 2 y Omer Kalayci, MD1
Resumen: Las especies reactivas de oxígeno (ROS) son producidas por organismos vivos como resultado del metabolismo celular normal y factores ambientales, como los contaminantes del aire o el humo del cigarrillo. Los ROS son moléculas altamente reactivas y pueden dañar estructuras celulares tales como carbohidratos, ácidos nucleicos, lípidos y proteínas y alterar sus funciones. El cambio en el equilibrio entre oxidantes y antioxidantes a favor de los oxidantes se denomina "estrés oxidativo". La regulación del estado reductor y oxidante (redox) es crítica para la viabilidad celular, la activación, la proliferación y la función del órgano. Los organismos aerobios tienen sistemas antioxidantes integrados, que incluyen antioxidantes enzimáticos y no enzimáticos que suelen ser eficaces para bloquear los efectos nocivos de los ROS. Sin embargo, en condiciones patológicas, los sistemas antioxidantes pueden ser abrumados. El estrés oxidativo contribuye a muchas condiciones y enfermedades patológicas, incluyendo cáncer, trastornos neurológicos, aterosclerosis, hipertensión, isquemia / perfusión, diabetes, síndrome de dificultad respiratoria aguda, fibrosis pulmonar idiopática, enfermedad pulmonar obstructiva crónica y asma. En esta revisión, resumimos los sistemas oxidantes y antioxidantes celulares y discutimos los efectos celulares y los mecanismos del estrés oxidativo.
Palabras clave: antioxidante, oxidante, estrés oxidativo, especies reactivas de oxígeno, redox
(Diario WAO 2012; 5: 9-19)
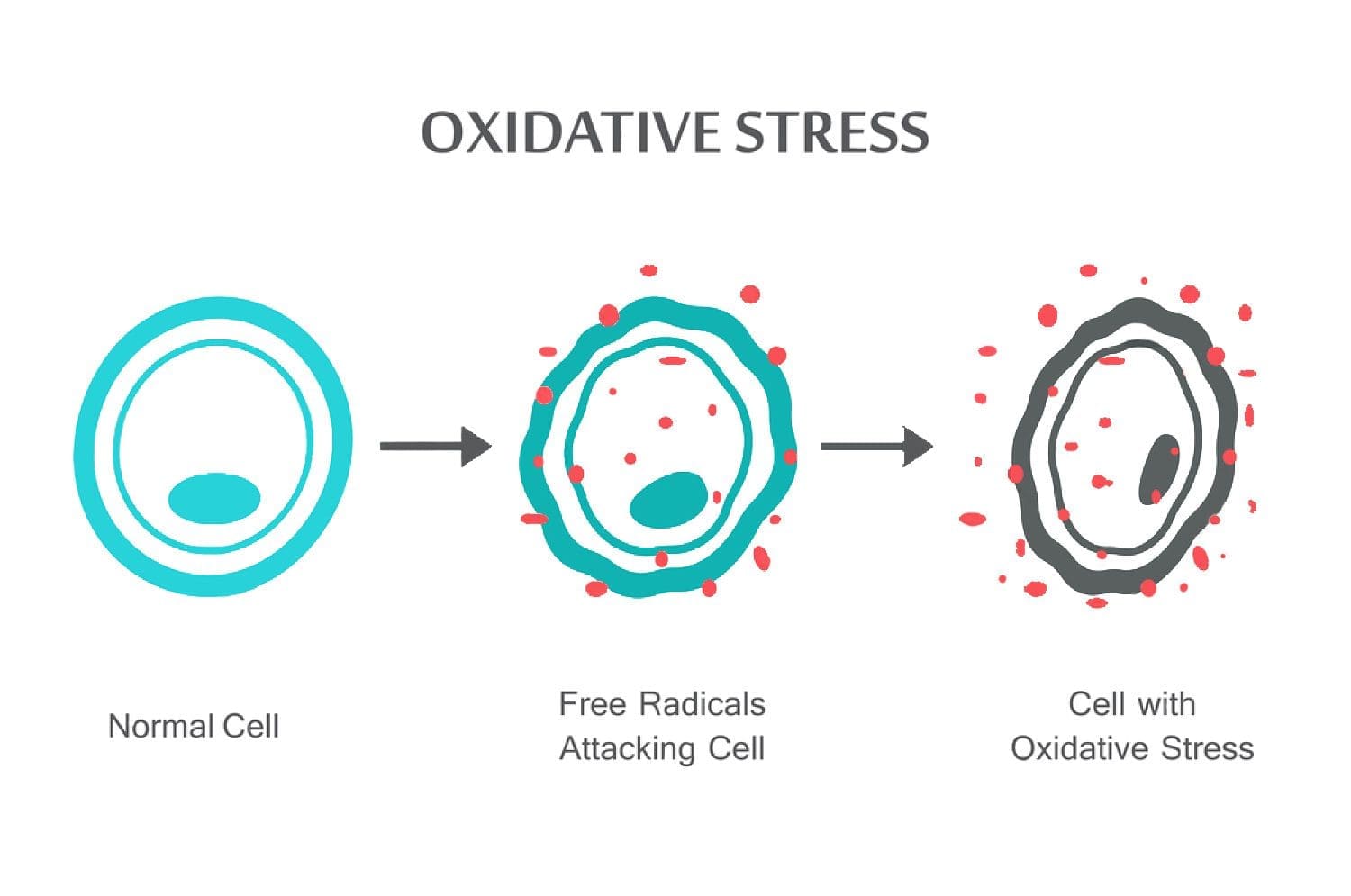
Las especies reactivas de oxígeno (ROS) son producidas por organismos vivos como resultado del metabolismo celular normal. En concentraciones bajas a moderadas, funcionan en procesos celulares fisiológicos, pero en concentraciones altas, producen modificaciones adversas en componentes celulares, como lípidos, proteínas y ADN.1-6 El cambio en el equilibrio entre oxidante / antioxidante a favor de oxidantes se denomina "estrés oxidativo". El estrés oxidativo contribuye a muchas afecciones patológicas, que incluyen cáncer, trastornos neurológicos, 7-10 aterosclerosis, hipertensión, isquemia / perfusión, 11-14 diabetes, síndrome de dificultad respiratoria aguda, fibrosis pulmonar idiopática, enfermedad pulmonar obstructiva crónica, 15 y asma16–. 21 Los organismos aeróbicos tienen sistemas antioxidantes integrados, que incluyen antioxidantes enzimáticos y no enzimáticos que suelen ser eficaces para bloquear los efectos dañinos de las ROS. Sin embargo, en condiciones patológicas, los sistemas antioxidantes pueden verse abrumados. En esta revisión, resumimos los sistemas oxidantes y antioxidantes celulares y la regulación del estado reductor y oxidante (redox) en estados de salud y enfermedad.
Índice del contenido
OXIDANTES
Fuentes endógenas de ROS
Los ROS se producen a partir de oxígeno molecular como resultado del metabolismo celular normal. Los ROS se pueden dividir en grupos 2: radicales libres y no radicales. Las moléculas que contienen uno o más electrones no emparejados y dando así reactividad a la molécula se llaman radicales libres. Cuando los radicales libres 2 comparten sus electrones no apareados, se crean formas no radicales. Los 3 principales ROS que tienen importancia fisiológica son el anión superóxido (O22.), El radical hidroxilo (OH) y el peróxido de hidrógeno (H2O2). ROS se resumen en la Tabla 1.
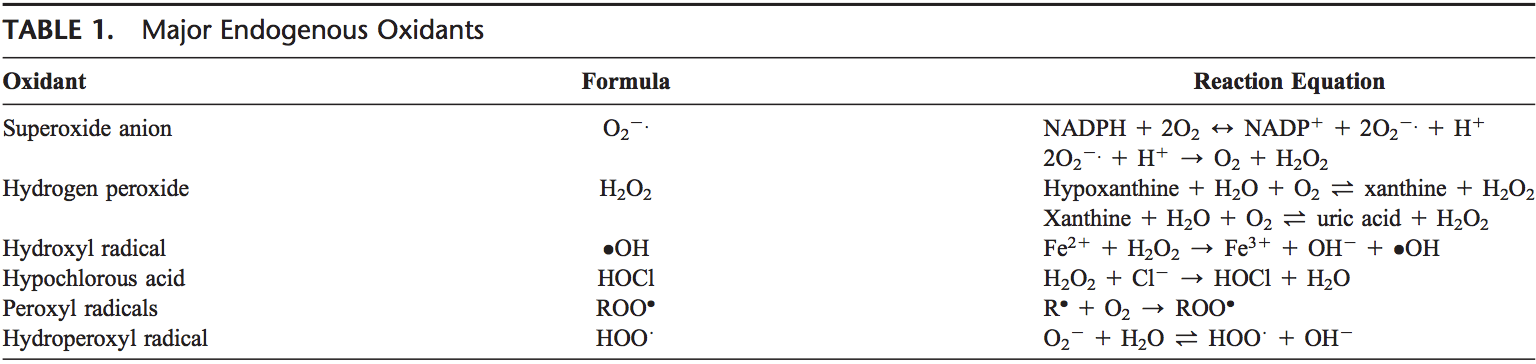
El anión superóxido se forma mediante la adición de 1 electrón al oxígeno molecular.22 Este proceso está mediado por el fosfato de dinucleótido de nicotina adenina [NAD (P) H] oxidasa o xantina oxidasa o por el sistema de transporte de electrones mitocondrial. El sitio principal de producción del anión superóxido son las mitocondrias, la maquinaria de la célula para producir trifosfato de adenosina. Normalmente, los electrones se transfieren a través de la cadena de transporte de electrones mitocondrial para reducir el oxígeno a agua, pero aproximadamente del 1 al 3% de todos los electrones se escapan del sistema y producen superóxido. La NAD (P) H oxidasa se encuentra en leucocitos polimorfonucleares, monocitos y macrófagos. Tras la fagocitosis, estas células producen una explosión de superóxido que conduce a la actividad bactericida. El superóxido se convierte en peróxido de hidrógeno por la acción de superóxido dismutasas (SOD, EC 1.15.1.1). El peróxido de hidrógeno se difunde fácilmente a través de la membrana plasmática. El peróxido de hidrógeno también es producido por la xantina oxidasa, el aminoácido oxidasa y la NAD (P) H oxidasa 23,24 y en los peroxisomas por el consumo de oxígeno molecular en reacciones metabólicas. En una sucesión de reacciones llamadas reacciones de Haber-Weiss y Fenton, el H2O2 puede descomponerse en OH2 en presencia de metales de transmisión como Fe21 o Cu21.25
Fe31 + .O2? Fe2 + O2 Haber Weiss
Fe2 + H2O2? Fe3 + OH + .OH Reacción de Fenton
El propio O 2 también puede reaccionar con H2 O2 y generar OH. 26,27 El radical hidroxilo es el más reactivo de los ROS y puede dañar proteínas, lípidos, carbohidratos y ADN. También puede iniciar la peroxidación de lípidos tomando un electrón de ácidos grasos poliinsaturados.
Las enzimas granulocíticas expanden aún más la reactividad de H2O2 a través de la peroxidasa eosinófila y la mieloperoxidasa (MPO). En los neutrófilos activados, H2O2 es consumido por MPO. En presencia de ion cloruro, H2O2 se convierte en ácido hipocloroso (HOCl). HOCl es altamente oxidativo y juega un papel importante en la muerte de los patógenos en las vías respiratorias. 28 Sin embargo, HOCl también puede reaccionar con el ADN e inducir las interacciones ADN-proteína y producir productos de oxidación pirimidina y añadir cloruro a las bases de ADN.29,30 Eosinophil peroxidase y MPO también contribuyen al estrés oxidativo mediante la modificación de proteínas mediante halogenaciones, nitración y enlaces cruzados de proteínas a través de radicales tirosilo. 31-33
Otros radicales libres derivados del oxígeno son los radicales peroxilo (ROO $). La forma más simple de estos radicales es el radical hidroperoxil (HOO $) y tiene un papel en la peroxidación de ácidos grasos. Los radicales libres pueden desencadenar reacciones en cadena de la peroxidación lipídica mediante la extracción de un átomo de hidrógeno de un carbono metileno de cadena lateral. El radical lipídico reacciona con el oxígeno para producir el radical peroxilo. El radical peroxilo inicia una reacción en cadena y transforma ácidos grasos poliinsaturados en hidroperóxidos lipídicos. Los hidroperóxidos de lípidos son muy inestables y se descomponen fácilmente en productos secundarios, tales como aldehídos (tales como 4-hidroxi-2,3-nonenal) y malondialdehídos (MDA). Los isoprostanos son otro grupo de productos de peroxidación de lípidos que se generan a través de la peroxidación del ácido araquidónico y también se ha encontrado que están elevados en el plasma y condensados de asma de los asmáticos.34,35 La peroxidación de lípidos perturba la integridad de las membranas celulares y conduce al reordenamiento de la estructura de membrana .
El peróxido de hidrógeno, el radical superóxido, el glutatión oxidado (GSSG), los MDA, los isoprostanos, los carbonilos y la nitrotirosina se pueden medir fácilmente a partir de muestras de lavado de plasma, sangre o broncoalveolar como biomarcadores de oxidación mediante ensayos estandarizados.
Fuente Exógena de Oxidantes
Humo de cigarro
El humo del cigarrillo contiene muchos oxidantes y radicales libres y compuestos orgánicos, como el superóxido y el óxido nítrico. Además, la inhalación de humo de cigarrillo en el pulmón también activa algunos mecanismos endógenos, como la acumulación de neutrófilos y macrófagos, lo que aumenta aún más la lesión oxidante .
Exposición al ozono
La exposición al ozono puede causar peroxidación de lípidos e inducir la afluencia de neutrófilos al epitelio de las vías respiratorias. La exposición a corto plazo al ozono también causa la liberación de mediadores inflamatorios, como la MPO, las proteínas catiónicas eosinófilas y también la lactato deshidrogenasa y la albúmina. 37 Incluso en sujetos sanos, la exposición al ozono causa una reducción de las funciones pulmonares. 38 Cho et al39 han demostrado que partículas (mezcla de partículas sólidas y gotas líquidas suspendidas en el aire) cataliza la reducción del oxígeno.
Hiperoxia
La hiperoxia se refiere a condiciones de niveles de oxígeno más altos que la presión parcial normal de oxígeno en los pulmones u otros tejidos corporales. Conduce a una mayor producción de especies reactivas de oxígeno y nitrógeno.40,41
Radiación ionizante
La radiación ionizante, en presencia de O2, convierte el radical hidroxilo, el superóxido y los radicales orgánicos en peróxido de hidrógeno e hidroperóxidos orgánicos. Estas especies de hidroperóxidos reaccionan con iones metálicos activos redox, tales como Fe y Cu, a través de reacciones de Fenton y por lo tanto inducen estrés oxidativo. 42,43 Narayanan et al44 demostró que los fibroblastos que estaban expuestos a partículas alfa tuvieron incrementos significativos en O2 2 intracelular. y la producción de H2O2 a través de la NADPH oxidasa ligada a la membrana plasmática. 44 Las moléculas de transducción de señales, tales como la quinasa 1 y 2 (ERK1 / 2), la quinasa N-terminal (JNK) y p38, y los factores de transcripción extracelulares, tales como la proteína activadora 1 (AP-1), el factor nuclear-kB (NF-kB) y p53, que resultan en la expresión de los genes relacionados con la respuesta a la radiación.45-50 Los fotones ultravioleta A (UVA) reacciones oxidativas por excitación de fotosensibilizadores endógenos, tales como porfirinas, NADPH oxidasa y riboflavinas. 8-Oxo-7,8-dihidroguanina (8-oxoGua) es el principal producto de oxidación del ADN mediado por UVA formado por la oxidación del radical OH, los oxidantes 1-electrón y el oxígeno singlete que reacciona principalmente con la guanina.51 La formación del radical radical guanina en el ADN aislado se ha demostrado que se producen eficazmente a través del efecto directo de la radiación ionizante. 52,53 Después de la exposición a la radiación ionizante, el nivel intracelular de glutatión (GSH) disminuye durante un corto plazo, pero luego aumenta de nuevo.
Iones de metales pesados
Los iones de metales pesados, como el hierro, el cobre, el cadmio, el mercurio, el níquel, el plomo y el arsénico, pueden inducir la generación de radicales reactivos y causar daño celular por agotamiento de las actividades enzimáticas a través de la peroxidación lipídica y la reacción con proteínas nucleares y ADN.
Uno de los mecanismos más importantes de la generación de radicales libres mediada por metales es a través de una reacción de tipo Fenton. El ion superóxido y el peróxido de hidrógeno pueden interactuar con metales de transición, como hierro y cobre, a través de la reacción de Haber-Weiss / Fenton catalizada por metal para formar radicales OH.
Metal31 1 $ O2 / Metal21 1 O2 Haber Weiss Metal21 1 H2 O2 / Metal31 1 OH 2 1 $ OH Reacción de Fenton
Además de los mecanismos tipo Fenton y Haber-Weiss, ciertos iones metálicos pueden reaccionar directamente con las moléculas celulares para generar radicales libres, como los radicales tiol, o inducir vías de señalización celular. Estos radicales también pueden reaccionar con otras moléculas tiol para generar O22 .. O22. se convierte en H2O2, lo que provoca una generación adicional de radicales de oxígeno. Algunos metales, como el arsenito, inducen indirectamente la formación de ROS mediante la activación de sistemas productores de radicales en las células.56
El arsénico es un elemento altamente tóxico que produce una variedad de ROS, incluyendo superóxido (O2 2), oxígeno singlete (1O2), radical peroxilo (ROO), óxido nítrico (NO), peróxido de hidrógeno (H2O2) y radicales peroxilo dimetilarsínico [ Los compuestos de arsénico (III) pueden inhibir las enzimas antioxidantes, especialmente las enzimas dependientes de GSH, tales como glutatión-S-transferasas (GSTs), glutatión peroxidasa (GSH-Px) y GSH reductasa, a través de enlaces - ing a sus grupos sulfhidrilo (-SH). 3
El plomo incrementa la peroxidación lipídica. 62 Se han reportado disminuciones significativas en la actividad de la SOD de tejidos y del cerebro GPx después de la exposición al plomo. 63,64 El reemplazo del zinc, que actúa como cofactor para muchas enzimas por plomo, conduce a la inactivación de tales enzimas. La exposición al plomo puede causar la inhibición del GST al afectar a los tioles del tejido.
Los ROS generados por reacciones catalizadas con metal pueden modular las bases de ADN. Tres sustituciones de la base, G / C, G / T y C / T, pueden ocurrir como resultado del daño oxidativo por los iones metálicos, como Fe21, Cu21 y Ni21. Reid et al65 demostraron que el G / C fue producido predominantemente por Fe21 mientras que la sustitución C / T fue por Cu21 y Ni21.
ANTIOXIDANTES
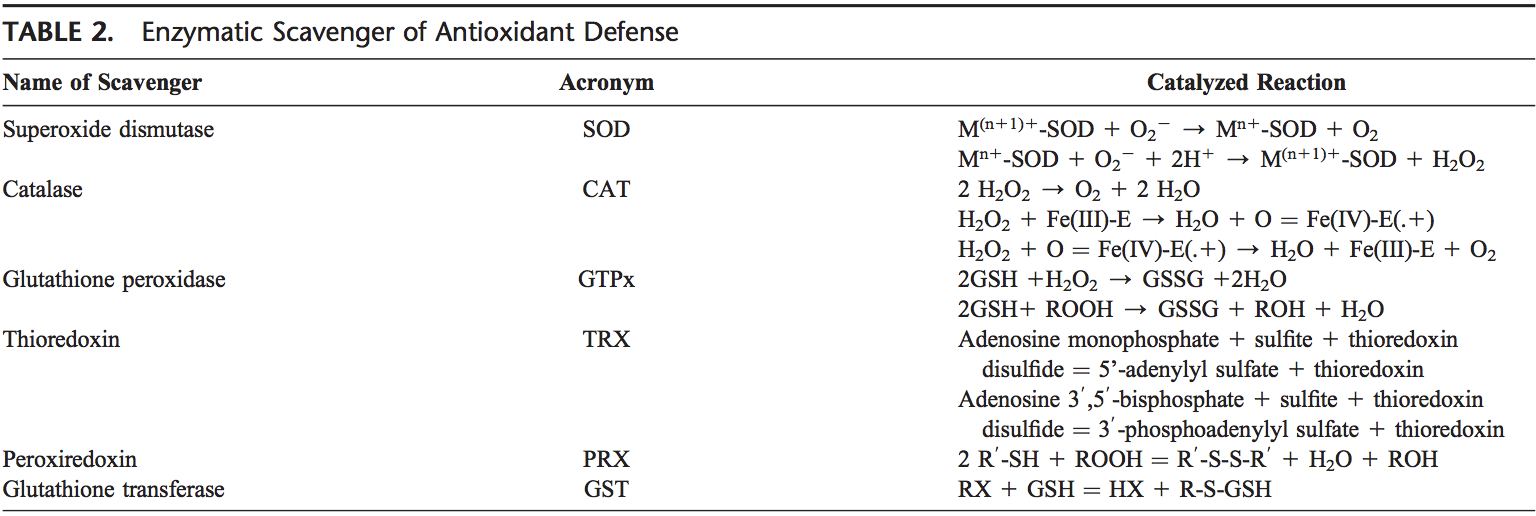
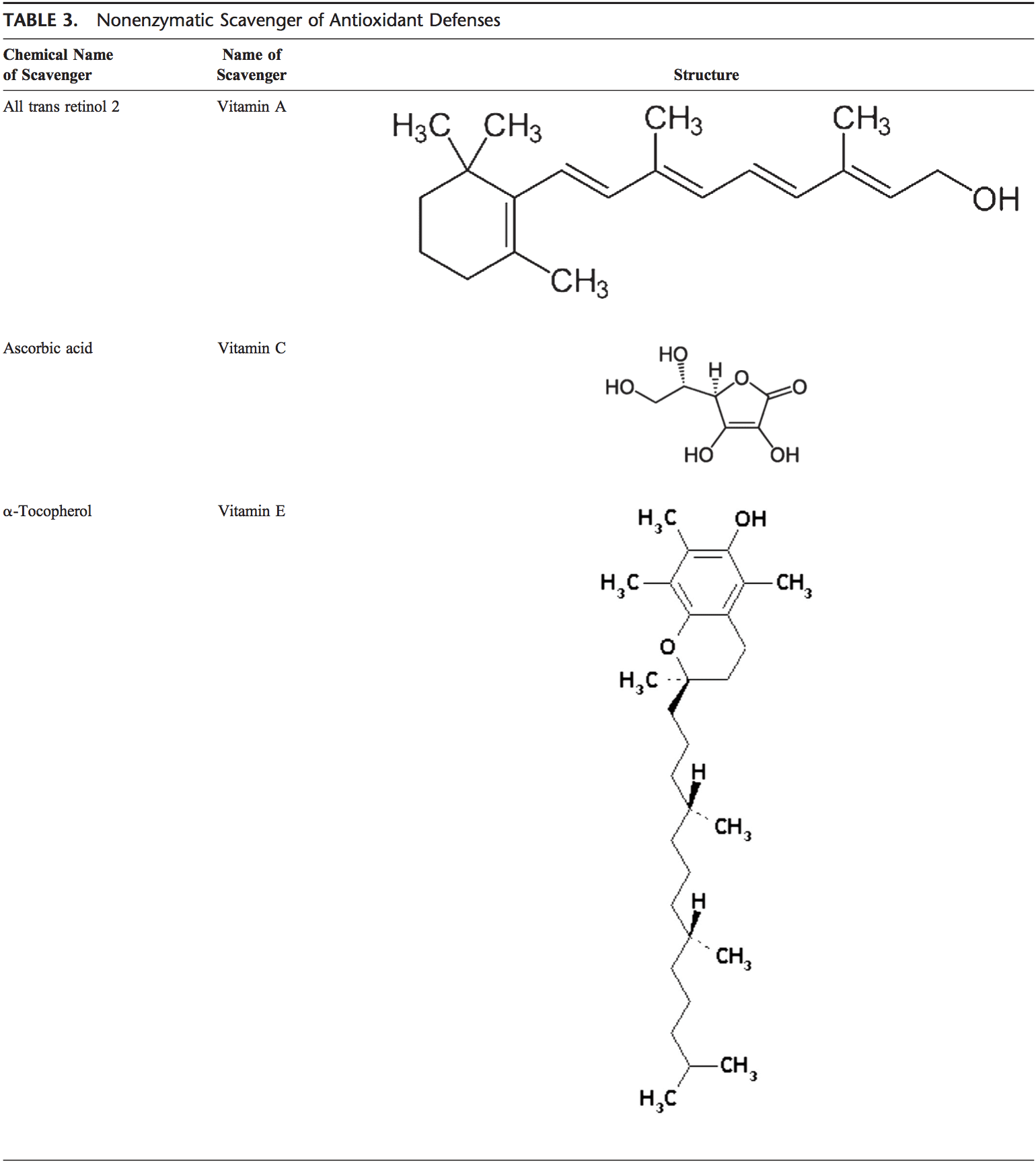
El cuerpo humano está equipado con una variedad de antioxidantes que sirven para contrarrestar el efecto de los oxidantes. Para todos los propósitos prácticos, estos pueden ser divididos en categorías 2: enzimática (Tabla 2) y no enzimática (Tabla 3).
Antioxidantes Enzimáticos
Los principales antioxidantes enzimáticos de los pulmones son SOD (EC 1.15.1.11), catalasa (EC 1.11.1.6) y GSH-Px (EC 1.11.1.9). Además de estas enzimas principales, se han encontrado también otros antioxidantes, incluyendo la oximexina-1 (EC 1.14.99.3) y las proteínas redox, tales como tiorredoxinas (TRXs, EC 1.8.4.10), peroxiredoxinas (PRXs, EC 1.11.1.15) y glutaredoxinas juegan un papel crucial en las defensas antioxidantes pulmonares.
Dado que el superóxido es el ROS primario producido a partir de una variedad de fuentes, su dismutación por SOD es de importancia primordial para cada célula. Todas las formas 3 de SOD, es decir, CuZn-SOD, Mn-SOD y EC-SOD, se expresan ampliamente en el pulmón humano. Mn-SOD se localiza en la matriz mitocondrial. EC-SOD se localiza principalmente en la matriz extracelular, especialmente en áreas que contienen altas cantidades de fibras de colágeno tipo I y alrededor de vasos pulmonares y sistémicos. También se ha detectado en el epitelio bronquial, el epitelio alveolar y los macrófagos alveolares. 66,67 En general, se piensa que CuZn-SOD y Mn-SOD actúan como depuradores a granel de radicales superóxido. El nivel EC-SOD relativamente alto en el pulmón con su unión específica a los componentes de la matriz extracelular puede representar un componente fundamental de la protección de la matriz pulmonar. 68
H2O2 que se produce por la acción de SODs o la acción de oxidasas, como la xantina oxidasa, se reduce a agua por la catalasa y el GSH-Px. La catalasa existe como un tetramero compuesto de monómeros idénticos 4, cada uno de los cuales contiene un grupo heme en el sitio activo. La degradación de H2O2 se realiza mediante la conversión entre las conformaciones 2 de catalasa-ferricatalasa (hierro coordinado al agua) y compuesto I (hierro complejado con un átomo de oxígeno). La catalasa también se une al NADPH como un equivalente reductor para evitar la inactivación oxidativa de la enzima (formación del compuesto II) por H2O2 a medida que se reduce a agua.69
Las enzimas del ciclo redox responsables de la reducción de H2O2 y de los hidroperóxidos lipídicos (generados como resultado de la peroxidación lipídica de la membrana) incluyen el GSH-Pxs.70 Los GSH-Px son una familia de enzimas tetraméricas que contienen la única selenocisteína de aminoácidos dentro del activos y utilizan tioles de bajo peso molecular, tales como GSH, para reducir H2O2 y peróxidos lipídicos a sus correspondientes alcoholes. Se han descrito cuatro GSH-Pxs, codificados por diferentes genes: GSH-Px-1 (GSH-Px celular) es omnipresente y reduce H2O2 y peróxidos de ácidos grasos, pero no los peroxil-lípidos esterificados.71 Los lípidos esterificados se reducen por GSH -Px-4 (hidroperóxido de fosfolípido GSH-Px), que puede utilizar varios tioles de bajo peso molecular como equivalentes reductores. GSH-Px-2 (GSH-Px extracelular) es el único miembro de la familia de GSH-Px que reside en las células epiteliales gastrointestinales. el compartimento extracelular y se cree que es una de las enzimas antioxidantes extracelulares más importantes en los mamíferos. De estos, GSH-Px extracelular es el más ampliamente investigado en el pulmón humano.72
Además, la eliminación de H2O2 está estrechamente asociada con varias enzimas que contienen tiol, a saber, las TRX (TRX1 y TRX2), las reductasas de la tiorredoxina (EC1.8.1.9) (TRRs), PRXs (que son peroxidasas de la tiorredoxina) y las glutaredoxinas.74
Dos TRXs y TRRs se han caracterizado en células humanas, existentes en citosol y mitocondrias. En el pulmón, TRX y TRR se expresan en epitelio bronquial y alveolar y macrófagos. Seis diferentes PRXs se han encontrado en las células humanas, que difieren en su compartimentación ultraestructural. Estudios experimentales han revelado la importancia de PRX VI en la protección del epitelio alveolar. El pulmón humano expresa todas las PRXs en el epitelio bronquial, el epitelio alveolar y los macrófagos. 75 PRX V recientemente se ha encontrado que funciona como una peroxinitrito reductasa, 76, lo que significa que puede funcionar como un potencial compuesto protector en el desarrollo de ROS mediada por lesión pulmonar .77
Común a estos antioxidantes es el requisito de NADPH como un equivalente reductor. NADPH mantiene la catalasa en forma activa y se utiliza como cofactor por TRX y GSH reductasa (EC 1.6.4.2), que convierte GSSG a GSH, un co-sustrato para el GSH-Pxs. El NADPH intracelular, a su vez, se genera por la reducción de NADP1 por la glucosa-6-fosfato deshidrogenasa, la primera enzima y limitante de la velocidad de la vía de pen- tosa fosfato, durante la conversión de fosfato de glucosa-6 a 6-fosfogluconolactona. Mediante la generación de NADPH, la glucosa-6-fosfato deshidrogenasa es un determinante crítico de la capacidad de amortiguación GSH citosólica (GSH / GSSG) y, por lo tanto, puede considerarse una enzima antioxidante reguladora esencial.78,79
Los GST (EC 2.5.1.18), otra familia de enzimas antioxidantes, inactivan los metabolitos secundarios, tales como aldehídos insaturados, epóxidos e hidroperóxidos. Se han descrito tres familias principales de GST: GST citosólico, GST mitocondrial, 80,81 y GST microsómico asociado a la membrana que tiene un papel en el metabolismo eicosanoide y GSH. 82 Se identifican siete clases de GST citosólicas en mamíferos, denominadas Alfa, Mu, Pi, Sigma, Theta, Omega y Zeta.83-86 Durante las condiciones no estresadas, la clase Mu y Pi GST interactúan con las quinasas Ask1 y JNK, respectivamente, e inhiben estas quinasas. 87-89 Se ha demostrado que GSTP1 se disocia de JNK en respuesta al estrés oxidativo.89 GSTP1 también interactúa físicamente con PRX VI y conduce a la recuperación de la actividad de la enzima PRX a través de glutationionación de la proteína oxidada.90
Antioxidantes no enzimáticos
Los antioxidantes no enzimáticos incluyen compuestos de bajo peso molecular, tales como vitaminas (vitaminas C y E), b-caroteno, ácido úrico y GSH, un tripéptido (Lg-glutamil-L-cisteinil-L-glicina) que comprende un tiol sulfhidrilo).
Vitamina C (ácido ascórbico)
La vitamina C soluble en agua (ácido ascórbico) proporciona capacidad antioxidante intracelular y extracelular de fase acuosa principalmente mediante la eliminación de radicales libres de oxígeno. Convierte los radicales libres de vitamina E en vitamina E. Se ha demostrado que sus niveles plasmáticos disminuyen con la edad. 91,92
Vitamina E (a-tocoferol)
La vitamina E soluble en lípidos se concentra en el sitio interior hidrofóbico de la membrana celular y es la principal defensa contra la lesión de la membrana inducida por el oxidante. La vitamina E dona electrones al radical peroxilo, que se produce durante la peroxidación de lípidos. El tocoferol es la forma más activa de la vitamina E y el principal antioxidante ligado a la membrana celular. La vitamina E desencadena la apoptosis de las células cancerosas e inhibe las formaciones de radicales libres.93
El glutatión
GSH es muy abundante en todos los compartimentos celulares y es el principal antioxidante soluble. GSH / GSSG es un factor determinante del estrés oxidativo. GSH muestra sus efectos antioxidantes de varias maneras.94 Desintoxica peróxido de hidrógeno y peróxidos lipídicos por acción de GSH-Px. GSH dona su electrón a H2O2 para reducirlo a H2O y O2. GSSG se reduce de nuevo en GSH por GSH reductasa que utiliza NAD (P) H como el donante de electrones. Los GSH-Px también son importantes para la protección de la membrana celular a partir de la peroxidación lipídica. El glutatión reducido dona protones a los lípidos de la membrana y los protege de los ataques de oxidantes. 95
GSH es un cofactor para varias enzimas desintoxicantes, tales como GSH-Px y transferasa. Tiene un papel en la conversión de la vitamina C y E de nuevo a sus formas activas. GSH protege las células contra la apoptosis mediante la interacción con vías de señalización proapoptóticas y antiapoptóticas. 94 También regula y activa varios factores de transcripción, como AP-1, NF-kB y Sp-1.
Carotenoides (b-caroteno)
Los carotenoides son pigmentos que se encuentran en las plantas. Principalmente, se ha encontrado que el b-caroteno reacciona con los radicales peroxilo (ROO), hidroxilo (OH) y superóxido (O22). 96 Los carotenoides muestran sus efectos antioxidantes en la baja presión parcial de oxígeno, pero pueden tener efectos prooxidantes en oxígeno superior concentraciones.97 Tanto los carotenoides como los ácidos retinoicos (RA) son capaces de regular factores de transcripción. 98 b-Caroteno inhibe la activación de NF-kB inducida por oxidante y la producción de interleucina (IL) -6 y factor de necrosis tumoral-a. Los carotenoides también afectan la apoptosis de las células. Los efectos antiproliferativos de la AR se han demostrado en varios estudios. Este efecto de la AR está mediado principalmente por receptores de ácido retinoico y varía entre los tipos de células. En las células de carcinoma mamario, se demostró que el receptor del ácido retinoico desencadenaba la inhibición del crecimiento induciendo el paro del ciclo celular, la apoptosis o ambos. 99,100
EL EFECTO DEL ESTRÉS OXIDATIVO: MECANISMOS GENÉTICOS, FISIOLÓGICOS Y BIOQUÍMICOS
El estrés oxidativo se produce cuando el equilibrio entre los antioxidantes y ROS se interrumpen debido a la depleción de antioxidantes o la acumulación de ROS. Cuando se produce un estrés oxidativo, las células intentan contrarrestar los efectos oxidantes y restaurar el equilibrio redox mediante la activación o el silenciamiento de genes que codifican enzimas defensivas, factores de transcripción y proteínas estructurales.101,102 La proporción entre glutatión oxidado y reducido (2GSH / GSSG) es una de los determinantes importantes del estrés oxidativo en el cuerpo. Una mayor producción de ROS en el cuerpo puede cambiar la estructura del ADN, resultar en la modificación de proteínas y lípidos, la activación de varios factores de transcripción inducida por el estrés, y la producción de pro-inflamatorios y anti-inflamatorios citoquinas.
Efectos del estrés oxidativo sobre el ADN
ROS puede conducir a modificaciones de ADN de varias maneras, lo que implica la degradación de bases, rupturas de DNA de una o dos cadenas, modificaciones, mutaciones, deleciones o translocaciones de purina, pirimidina o azúcar, y reticulación con proteínas. La mayoría de estas modificaciones del ADN (Fig. 1) son altamente relevantes para la carcinogénesis, el envejecimiento y las enfermedades neurodegenerativas, cardiovasculares y autoinmunes. El humo del tabaco, los metales redox y los metales no redox, como el hierro, el cadmio, el cromo y el arsénico, también están implicados en la carcinogénesis y el envejecimiento al generar radicales libres o unirse a grupos tiol. La formación de 8-OH-G es el daño ADN más conocido que se produce a través del estrés oxidativo y es un biomarcador potencial para la carcinogénesis.
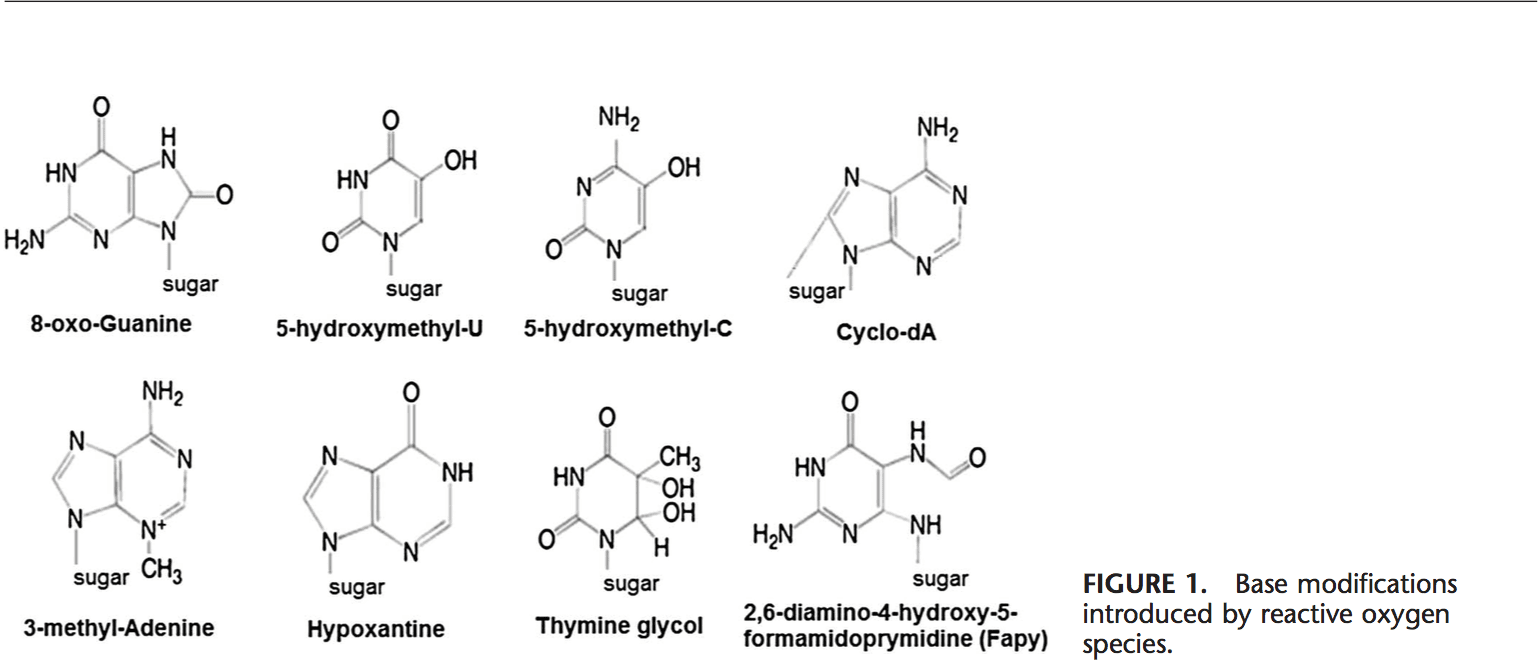
Las regiones promotoras de genes contienen secuencias de consenso para factores de transcripción. Estos sitios de unión al factor de transcripción contienen secuencias ricas en GC que son susceptibles de ataques oxidantes. La formación de ADN de 8-OH-G en sitios de unión de factor de transcripción puede modificar la unión de factores de transcripción y por lo tanto cambiar la expresión de genes relacionados como se ha demostrado para las secuencias diana de AP-1 y Sp-1. 103 Además de 8-OH- También se ha demostrado que la 8,59-desoxiadenosina (ciclo-dA) inhibe la transcripción de un gen reportero en un sistema celular si está localizada en una caja TATA.29 La proteína de unión a TATA inicia la transcripción cambiando la flexión del ADN. La unión de la proteína de unión a TATA puede verse afectada por la presencia de ciclo-dA.
El estrés oxidativo causa la inestabilidad de las regiones de microsatélites (repeticiones en tándem corto). Los iones de metal reactivos redox, los radicales hidroxilo aumentan la inestabilidad de los microsatélites. 105 Aunque las rupturas de ADN monocatenario causadas por lesiones oxidantes pueden ser fácilmente toleradas por las células, las rupturas de ADN bicatenario inducidas por radiación ionizante pueden ser una amenaza significativa para la supervivencia celular.
La metilación en las islas CpG en el ADN es un importante mecanismo epigenético que puede resultar en el silenciamiento de genes. Oxidación de 5-MeCyt a 5-hidroximetil uracilo (5-OHMeUra) puede ocurrir a través de reacciones de desaminación / oxidación de timina o 5-hidroximetil citosina intermedios.107 Además de la modulación de la expresión génica, la metilación del ADN también parece afectar la cromatina organization.108 Los patrones aberrantes de metilación del ADN inducidos por ataques oxidativos también afectan la actividad de reparación del ADN.
Efectos del estrés oxidativo sobre los lípidos
Los ROS pueden inducir peroxidación de lípidos e interrumpir la disposición de bicapa de membrana que puede inactivar los receptores y enzimas unidos a la membrana y aumentar la permeabilidad de los tejidos. 109 Los productos de la peroxidación lipídica, tales como MDA y aldehídos insaturados, son capaces de inactivar muchas proteínas celulares formando proteínas cruzadas El 110 activa el receptor del factor de crecimiento epidérmico, 112 e induce la producción de fibronectina. 4 Los productos de peroxidación de los lípidos, tales como las sustancias reactivas al ácido isoprostano y tiobarbitúrico , se han utilizado como biomarcadores indirectos del estrés oxidativo, y los niveles aumentados se mostraron en el condensado de aire exhalado o líquido de lavado broncoalveolar o pulmón de pacientes con enfermedad pulmonar obstructiva crónica o fumadores.2-113,114
Efectos del estrés oxidativo sobre las proteínas
ROS puede causar la fragmentación de la cadena peptídica, la alteración de la carga eléctrica de las proteínas, la reticulación de las proteínas y la oxidación de aminoácidos específicos y, por lo tanto, conducir a una mayor susceptibilidad a la proteólisis por degradación por proteasas específicas.120 Cisteína y metionina residuos en proteínas son particularmente más susceptibles a la oxidación.121 La oxidación de grupos sulfhidrilo o residuos de metionina de proteínas causan cambios conformacionales, despliegue de proteínas y degradación. 8,121-123 Las enzimas que tienen metales en sus sitios activos o cerca de ellos son especialmente más sensibles a la oxidación catalizada por metal. Se ha demostrado que la modificación oxidativa de las enzimas inhibe sus actividades.124,125
En algunos casos, puede tener lugar la oxidación específica de las proteínas. Por ejemplo, la metionina puede ser sulfóxido de metionina oxidado 126 y fenilalanina a o-tirosina 127; los grupos sulfhidrilo se pueden oxidar para formar enlaces disulfuro, 128 y grupos carbonilo pueden introducirse en las cadenas laterales de proteínas. Los rayos gamma, la oxidación catalizada por metales, el HOCl y el ozono pueden causar la formación de grupos carbonilo.129
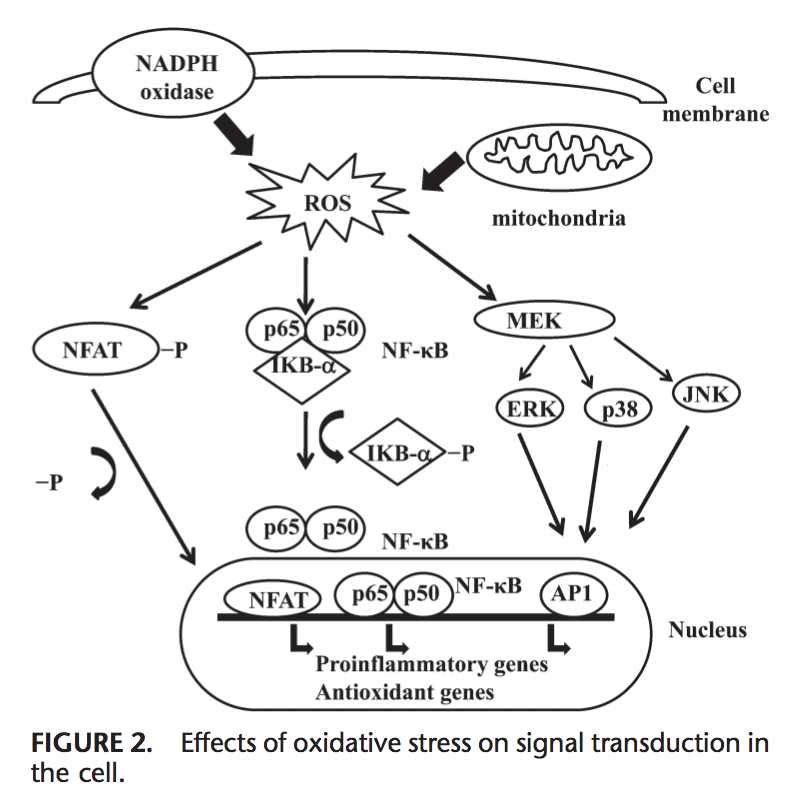
Efectos del estrés oxidativo sobre la transducción de señales
ROS puede inducir la expresión de varios genes implicados en la transducción de la señal. 1,130 Una alta proporción para GSH / GSSG es importante para la protección de la célula contra daño oxidativo. La interrupción de esta relación provoca la activación de factores de transcripción sensibles al redox, tales como NF-kB, AP-1, factor nuclear de células T activadas y factor 1 inducible por hipoxia, que están implicados en la respuesta inflamatoria. La activación de los factores de transcripción a través de ROS se logra mediante cascadas de transducción de señales que transmiten la información desde el exterior hacia el interior de la célula. Los receptores de tirosina quinasa, la mayoría de los receptores del factor de crecimiento, tales como el receptor del factor de crecimiento epidérmico, el receptor del factor de crecimiento endotelial vascular y el receptor para el factor de crecimiento derivado de plaquetas, las tirosina fosfatasas de proteínas y las quinasas serina / treonina son blancos de ROS.131-133 Las quinasas reguladas por señal extracelular, JNK y p38, que son miembros de la familia de proteínas quinasas activadas por mitógenos y que participan en varios procesos en la célula incluyendo proliferación, diferenciación y apoptosis, también pueden ser reguladas por oxidantes.
En condiciones de estrés oxidativo, los residuos de cisteína en el sitio de unión al ADN de c-Jun, algunas subunidades de AP-1 y kB quinasa inhibitoria experimentan S-glutatiolación reversible. Se ha informado que la glutaredoxina y la TRX desempeñan un papel importante en la regulación de vías de señalización sensibles a redox, tales como NF-kB y AP-1, la proteína quinasa activada por mitógeno p38 y JNK.134-137
NF-kB puede ser activado en respuesta a condiciones de estrés oxidativo, tales como ROS, radicales libres y radiación UV. 138 La fosforilación de IkB libera NF-kB y le permite entrar en el núcleo para activar la transcripción de genes.139 Un número de quinasas se ha informado que fosforila IkBs en los residuos de serina. Estas quinasas son los objetivos de las señales oxidativas para la activación de NF-kB.140 Los agentes reductores mejoran la unión al ADN de NF-kB, mientras que los agentes oxidantes inhiben la unión al ADN de NF-kB. TRX puede ejercer 2 acciones opuestas en la regulación de NF-kB: en el citoplasma, bloquea la degradación de IkB e inhibe la activación de NF-kB, pero mejora la unión del ADN de NF-kB en el núcleo.141 Activación de NF-kB a través de la degradación relacionada con la oxidación de IkB resulta en la activación de varios genes antioxidantes relacionados con la defensa. NF-kB regula la expresión de varios genes que participan en la respuesta inmune, tales como IL-1b, IL-6, factor de necrosis tumoral-a, IL-8 y varias moléculas de adhesión.142,143 NF-kB también regula angiogénesis y proliferación y diferenciación de células.
AP-1 también está regulado por el estado redox. En presencia de H2O2, algunos iones metálicos pueden inducir la activación de AP-1. El aumento de la relación de GSH / GSSG mejora la unión de AP-1 mientras que GSSG inhibe la unión de ADN de AP-1.144 La unión de ADN del heterodímero Fos / Jun se incrementa mediante la reducción de una única cisteína conservada en el dominio de unión a ADN de cada uno de las proteínas, 145 mientras que el ADN vinculante de AP-1 puede ser inhibido por GSSG en muchos tipos de células, lo que sugiere que la formación de enlaces disulfuro de residuos de cisteína inhibe AP-1 ADN binding.146,147 Transducción de señal a través de estrés oxidativo se resume en la Figura 2.
CONCLUSIONES
El estrés oxidativo puede surgir de la sobreproducción de ROS por las reacciones metabólicas que utilizan el oxígeno y cambiar el equilibrio entre oxidanteantioxidante estatus a favor de los oxidantes. Los ROS son producidos por actividades metabólicas celulares y factores ambientales, como los contaminantes del aire o el humo del cigarrillo. Los ROS son moléculas altamente reactivas debido a electrones no emparejados en su estructura y reaccionan con varias macromoléculas biológicas en la célula, tales como carbohidratos, ácidos nucleicos, lípidos y proteínas, y alteran sus funciones. ROS también afecta a la expresión de varios genes por upregulation de redox sensibles factores de transcripción y remodelación de la cromatina a través de la alteración en la acetilación / desacetilación de las histonas. La regulación del estado redox es crítica para la viabilidad celular, activación, proliferación y función de órganos.
Referencias:
Referencias
1. Valko M, Rodas CJ, Moncol J, Izakovic M, Mazur M. Radicales libres, metales y antioxidantes en el estrés oxidativo inducido por el cáncer. Chem Biol Interact. 2006; 160: 1-40.
2. Halliwell B, Gutteridge JMC. Radicales libres en biología y medicina. 3rd ed. Nueva York: Oxford University Press, 1999.
3. Marnett LJ. La peroxidación de los lípidosDNA daño por malondialdehído. Mutat Res. 1999; 424: 83-95.
4. Siems WG, Grune T, Esterbauer H. Formación de 4-hidroxinonenal durante la isquemia y reperfusión del intestino delgado de rata. Life Sci. 1995; 57: 785–789.
5. Stadtman ER. Papel de las especies oxidantes en el envejecimiento. Curr Med Chem. 2004; 11: 1105-1112.
6. Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. Aductos putativos malondialdehído-ADN inducidos por peroxidación de lípidos en tejidos mamarios humanos. Cáncer Epidemiol Biomarcadores Prev. 1996; 5: 705-710.
7. Jenner P. Estrés oxidativo en la enfermedad de Parkinson. Ann Neurol. 2003; 53: S26-S36.
8. Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. Una evaluación del daño oxidativo a proteínas, lípidos y ADN en el cerebro de pacientes con enfermedad de Alzheimer. J Neurochem. 1997; 68: 2061-2069.
9. Sayre LM, Smith MA, Perry G. Química y bioquímica del estrés oxidativo en las enfermedades neurodegenerativas. Curr Med Chem. 2001; 8: 721-738.
10. Toshniwal PK, Zarling EJ. Evidencia de aumento de la peroxidación lipídica en la esclerosis múltiple. Neurochem Res. 1992; 17: 205-207.
11. Dhalla NS, Temsah RM, Netticadan T. Papel del estrés oxidativo en las enfermedades cardiovasculares. J Hypertens. 2000; 18: 655-673.
12. Kasparova S, Brezova V, Valko M, Horecky J, Mlynarik V, et al. Estudio del estrés oxidativo en un modelo de rata de hipoperfusión cerebral crónica. Neurochem Int. 2005; 46: 601-611.
13. Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. La producción de aniones superóxido se incrementa en un modelo de hipertensión genética: papel del endotelio. Hipertensión. 1999; 33: 1353-1358.
14. Kukreja RC, Hess ML. El sistema de radicales libres de oxígeno: desde ecuaciones a través de interacciones membrana-proteína hasta lesiones cardiovasculares y protección. Cardiovasc Res. 1992; 26: 641-655.
15. Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T, et al. El tabaquismo induce un aumento en el daño oxidativo del ADN, 8-hydroxydeoxyguanosine, en un sitio central del pulmón humano. Carcinogénesis. 1997; 18: 1763-1766.
16. Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Eventos oxidativos y nitrosativos en el asma. Gratis Radic Biol Med. 2003; 35: 213-225.
17. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA, et al. Correlación de la deficiencia sistémica de superóxido dismutasa con la obstrucción del flujo aéreo en el asma. Am J Respir Crit Care Med. 2005; 172: 306-313.
18. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A, et al. Inactivación de la superóxido dismutasa en la fisiopatología de la remodelación asmática de las vías aéreas y su reactividad. Am J Pathol. 2005; 166: 663-674.
19. Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. El estrés oxidativo y sus determinantes en las vías respiratorias de los niños con asma. Alergia. 2008; 63: 1605-1609.
20. Ercan H, Birben E, Dizdar EA, Keskin O, Karaaslan C, y col. Estrés oxidativo y determinantes genéticos y epidemiológicos de la lesión oxidante en el asma infantil. J Allergy Clin Immunol. 2006; 118: 1097-1104.
21. Fitzpatrick AM, GT Teague, Holguin F, Yeh M, Brown LA. Programa de Investigación de Asma Grave. La homeostasis de glutatión en las vías respiratorias está alterada en niños con asma grave: evidencia de estrés oxidante. J Allergy Clin Immunol. 2009; 123: 146-152.
22. Miller DM, Buettner GR, Aust SD. Metales de transición como catalizadores de reacciones de "autoxidación". Gratis Radic Biol Med. 1990; 8: 95-108.
23. Dupuy C, Virion A, Ohayon R, Kaniewski J, Dème D, Pommier J. Mecanismo de formación de peróxido de hidrógeno catalizado por la NADPH oxidasa en la membrana plasmática tiroidea. J Biol Chem. 1991; 266: 3739-3743.
24. Granger DN. Papel de la xantina oxidasa y de los granulocitos en la lesión isquémica por perfusión. Am J Physiol. 1988; 255: H1269-H1275.
25. Fenton HJH. Oxidación del ácido tartárico en presencia de hierro. J Chem Soc. 1984; 65: 899-910.
26. Haber F, Juez Weiss. La descomposición catalítica del peróxido de hidrógeno por sales de hierro. Proc R Soc Lond Ser A. 1934; 147: 332-351.
27. Liochev SI, Fridovich I. El Haber-Weiss cycled70 años más tarde: una visión alternativa. Redox Rep. 2002; 7: 55-57.
28. Klebanoff SJ. Mieloperoxidasa: amigo y enemigo. J Leukoc Biol. 2005; 77: 598-625.
29. Whiteman M, Jenner A, Halliwell B. Alteraciones de base inducidas por el ácido hipocloroso en ADN aislado del timo de ternera. Chem Res Toxicol. 1997; 10: 1240-1246.
30. Kulcharyk PA, Heinecke JW. El ácido hipocloroso producido por el sistema mieloperoxidasa de los fagocitos humanos induce enlaces cruzados covalentes entre el ADN y la proteína. Bioquímica. 2001; 40: 3648-3656.
31. Brennan ML, Wu W, Fu X, Shen Z, Song W, y col. Un cuento de dos controversias: definir el papel de las peroxidasas en la formación de nitrotirosina in vivo utilizando peroxidasa eosinófilo y myeloperoxidas ratones deficientes, y la naturaleza de las especies de nitrógeno reactivo generado por peroxidasa. J Biol Chem. 2002; 277: 17415-17427.
32. Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM, et al. La desgranulación extensa de eosinófilos y la oxidación mediada por peroxidasa de las proteínas de las vías respiratorias no se producen en un modelo de inflamación pulmonar provocado por la ovoalbúmina del ratón. J Immunol. 2001; 167: 1672-1682.
33. CJ de van Dalen, Winterbourn CC, Senthilmohan R, Hervidor de agua AJ. Nitrito como sustrato e inhibidor de la mieloperoxidasa. Implicaciones para la producción de nitrato y ácido hipocloroso en sitios de inflamación. J Biol Chem. 2000; 275: 11638-11644.
34. Madera LG, Fitzgerald DA, Gibson PG, Cooper DM, Garg ML. La peroxidación lipídica determinada por los isoprostanos plasmáticos está relacionada con la gravedad de la enfermedad en el asma leve. Lípidos. 2000; 35: 967-974.
35. Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Kharitonov SA, Barnes PJ. Aumento de 8-isoprostano, un marcador de estrés oxidativo, en el condensado exhalado de pacientes con asma. Am J Respir Crit Care Med. 1999; 160: 216-220.
36. Iglesia DF, Pryor WA. Química de radicales libres del humo del cigarrillo y sus implicaciones toxicológicas. Perspectiva de salud ambiental. 1985; 64: 111-126.
37. Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ, et al. Home Idiomas Ingresar a Epistemonikos Búsqueda avanzada Inflamación inducida por ozono evaluado en el esputo y el líquido de lavado bronquial de asmáticos: una nueva herramienta no invasiva en estudios epidemiológicos sobre la contaminación del aire y el asma. Gratis Radic Biol Med. 1999; 27: 1448-1454.
38. Nightingale JA, Rogers DF, Barnes PJ. Efecto del ozono inhalado sobre el óxido nítrico exhalado, la función pulmonar y el esputo inducido en sujetos normales y asmáticos. Tórax. 1999; 54: 1061-1069.
39. Cho AK, Sioutas C, Miguel AH, Kumagai Y, Schmitz DA, et al. Actividad redox de partículas en suspensión en el aire en diferentes sitios de la Cuenca de Los Ángeles. Environ Res. 2005; 99: 40-47.
40. Comhair SA, Thomassen MJ, Erzurum SC. Inducción diferencial de glutatión peroxidasa extracelular y óxido nítrico sintasa 2 en vías respiratorias de individuos sanos expuestos a 100% O (2) o humo de cigarrillo. Am J Respir Cell Mol Biol. 2000; 23: 350-354.
41. Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Lesión pulmonar mediada por oxidante en el síndrome de dificultad respiratoria aguda. Crit Care Med. 1999; 27: 2028-2030.
42. Biaglow JE, Mitchell JB, Held K. La importancia del peróxido y el superóxido en la respuesta de rayos X. Int J Radiat Oncol Biol Phys. 1992; 22: 665-669.
43. Chiu SM, Xue LY, Friedman LR, Oleinick NL. Sensibilización mediada por iones de cobre de los sitios de fijación de la matriz nuclear a la radiación ionizante. Bioquímica. 1993; 32: 6214-6219.
44. Narayanan PK, Goodwin EH, Lehnert BE. Las partículas alfa inician la producción biológica de aniones superóxido y peróxido de hidrógeno en células humanas. Cancer Res. 1997; 57: 3963-3971.
45. Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Sensibilidad a oxidantes químicos y radiación en líneas celulares CHO deficientes en la actividad oxidativa del ciclo de las pentosas. Int J Radiat Oncol Biol Phys. 1992; 22: 671-675.
46. Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D, y col. Manganeso
superóxido dismutasa mediada por la expresión génica en radiación inducida por
respuestas adaptativas. Mol Cell Biol. 2003; 23: 2362-2378.
47. Azzam EI, de Toledo SM, Spitz DR, Little JB. Metabolismo oxidativo
modula la transducción de señales y la formación de micronúcleos en espectadores
células de fibroblastos humanos normales irradiados con una partícula. Cancer Res.
2002; 62: 5436-5442.
48. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Generación ionizante inducida por radiación, mitocondria-dependiente de reactivo
oxígeno / nitrógeno. Cancer Res. 2001; 61: 3894-3901.
49. Dent P, Yacoub A, Fisher PB, MP Hagan, Grant S. MAPK
radiación. Oncogene. 2003; 22: 5885-5896.
50. Wei SJ, Botero A, Hirota K, CM Bradbury, Markovina S, y col. Thioredoxin
la translocación nuclear y la interacción con el factor redox 1 activa el factor de transcripción AP-1 en respuesta a la radiación ionizante. Cancer Res. 2000; 60: 6688-6695.
51. Cadet J, Douki T, Gasparutto D, Ravanat JL. Daño oxidativo al ADN: formación, medida y características bioquímicas. Mutat Res. 2003; 531: 5-23.
52. Yokoya A, Cunniffe SM, O'Neill P. Efecto de la hidratación en la inducción de rupturas de la cadena y lesiones de base en películas de ADN plasmídico mediante gammaradiación. J Am Chem Soc. 2002; 124: 8859-8866.
53. Janssen YM, Van Houten B, Borm PJ, Mossman BT. Respuesta de células y tejidos al daño oxidativo. Lab Invest. 1993; 69: 261-274.
54. Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T, et al. Factor nuclear kappa B que induce la inducción de gamma glutamilcisteína sintetasa por radiación ionizante en células de glioblastoma humano T98G. Gratis Radic Biol Med. 1998; 24: 1256-1268.
55. Stohs SJ, Bagchi D. Mecanismos oxidativos en la toxicidad de los iones metálicos. Gratis Radic Biol Med. 1995; 18: 321-336.
56. Leonard SS, Harris GK, Shi X. Estrés oxidativo inducido por el metal y transducción de señales. Gratis Radic Biol Med. 2004; 37: 1921-1942.
57. Shi H, Shi X, Liu KJ. Mecanismo oxidativo de toxicidad por arsénico y carcinogénesis. Mol Cell Biochem. 2004; 255: 67-78.
58. Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. Un mecanismo potencial para el deterioro de la formación de óxido nítrico causada por la exposición oral prolongada al arseniato en conejos. Radic Biol gratis Med.2003; 35: 102-113.
59. Rin K, Kawaguchi K, Yamanaka K, Tezuka M, Oku N, Okada S. Las rupturas de DNAstrand inducidas por el ácido dimetilarsinico, un metabolito de arsénicos inorgánicos, son fuertemente potenciadas por los radicales anión superóxido. Biol Pharm Bull. 1995; 18: 45-58.
60. Waalkes MP, Liu J, Ward JM, Diwan LA. Mecanismos subyacentes a la carcinogénesis de arsénico: hipersensibilidad de ratones expuestos al arsénico inorgánico durante la gestación. Toxicología. 2004; 198: 31-38.
61. Schiller CM, Fowler BA, Woods JS. Efectos del arsénico sobre la activación de piruvato deshidrogenasa. Perspectiva de salud ambiental. 1977; 19: 205-207.
62. Monterio HP, Bechara EJH, Abdalla DSP. Participación de radicales libres en porfirias neurológicas y envenenamiento por plomo. Mol Cell Biochem. 1991; 103: 73-83.
63. Tripathi RM, Raghunath R, Mahapatra S. Plomo en sangre y su efecto sobre los niveles de Cd, Cu, Zn, Fe y hemoglobina de los niños. Sci Total Environ. 2001; 277: 161-168.
64. Nehru B, Dua R. El efecto del selenio en la dieta sobre la neurotoxicidad del plomo. J Environ Patol Toxicol Oncol. 1997; 16: 47-50.
65. Reid TM, Feig DI, Loeb LA. Mutagénesis por radicales de oxígeno inducidos por metal. Perspectiva de salud ambiental. 1994; 102 (suplemento 3): 57-61.
66. Kinnula VL, Crapo JD. Superóxido dismutasas en el pulmón y enfermedades pulmonares humanas. Am J Respir Crit Care Med. 2003; 167: 1600-1619.
67. Kinnula VL. Producción y degradación de metabolitos de oxígeno durante estados inflamatorios en el pulmón humano. El Diario Curr Dirige Alergia Inflamatoria. 2005; 4: 465-470.
68. Zelko IN, Mariani TJ, Folz RJ. Home Idiomas Ingresar a Epistemonikos Búsqueda avanzada Familia multigénica de superóxido dismutasa: una comparación de las estructuras génicas, evolución y expresión de los genes CuZn-SOD (SOD1), Mn-SOD (SOD2) y EC-SOD (SOD3). Gratis Radic Biol Med. 2002; 33: 337-349.
69. Kirkman HN, Rolfo M, Ferraris AM, Gaetani GF. Mecanismos de protección de catalasa por NADPH. Cinética y estequiometría. J Biol Chem. 1999; 274: 13908-13914.
70. Flohé L. Glutatión peroxidasa. Vida Básica Sci. 1988; 49: 663-668.
71. Arthur JR. Las glutatión peroxidasas. Cell Mol Life Sci. 2000; 57: 1825-1835.
72. Chu FF, Doroshow JH, Esworthy RS. Expresión, caracterización y distribución tisular de una nueva glutation peroxidasa dependiente de selenio celular, GSHPx-GI. J Biol Chem. 1993; 268: 2571-2576.
73. Comhair SA, Bhathena PR, C Farver, Thunnissen FB, Erzurum SC. Inducción extracelular de glutatión peroxidasa en los pulmones asmáticos: evidencia de la regulación redox de la expresión en células epiteliales de las vías respiratorias humanas. FASEB J. 2001; 15: 70-78.
74. Gromer S, Urig S, Becker K. El sistema de la tiorredoxina de la ciencia a la clínica. Med Rev Rev. 2004; 24: 40-89.
75. Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, Pääkkö P, et al. Expresión celular específica de peroxiredoxinas en pulmón humano y sarcoidosis pulmonar. Tórax. 2002; 57: 157-164.
76. Dubuisson M, Vander Stricht D, Clippe A, Etienne F., Nauser T, et al. La peroxiredoxina humana 5 es una peroxinitrito reductasa. FEBS Lett. 2004; 571: 161-165.
77. Holmgren A. Función antioxidante de los sistemas de tiorredoxina y glutaredoxina. Señal Redox De Antioxid. 2000; 2: 811-820.
78. Dickinson DA, Forman HJ. Glutatión en defensa y señalización: lecciones de un pequeño tiol. Ann NY Acad Sci. 2002; 973: 488-504.
79. Sies H. Glutathione y su papel en las funciones celulares. Gratis Radic Biol Med. 1999; 27: 916-921.
80. Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Home Cómo funciona Idiomas Ingresar a Epistemonikos Búsqueda avanzada Caminos evolutivos paralelos para las transfertasas de glutatión: estructura y mecanismo de la enzima kappa de la clase mitocondrial rGSTK1-1. Bioquímica. 2004; 43: 52-61.
81. Robinson A, Huttley GA, Stand HS, Junta PG. Los estudios de modelado y bioinformática de la glutatión transferasa de clase kappa humana predicen una tercera familia de tercera transferasa con homología con 2-hidroxicromeno-2-carboxilato isomerasas procarióticas. Biochem J. 2004; 379: 541 - 552.
82. Jakobsson PJ, Morgenstern R, Mancini J., Ford-Hutchinson A, Persson B. Características estructurales comunes de MAPEGda superfamilia extendida de proteínas asociadas a la membrana con funciones altamente divergentes en el metabolismo eicosanoide y glutatión. Protein Sci. 1999; 8: 689-692.
83. Hayes JD, Pulford DJ. La familia de supergenes S-transferasa de glutatión: regulación de GST y la contribución de las isoenzimas a la quimioprotección y resistencia al fármaco. Crit Rev Biochem Mol Biol. 1995; 30: 445-600.
84. Armstrong RN. Estructura, mecanismo catalítico y evolución de las transferencias de glutatión. Chem Res Toxicol. 1997; 10: 2-18.
85. Hayes JD, McLellan LI. El glutatión y las enzimas dependientes del glutatión representan una defensa regulada de forma coordinada contra el estrés oxidativo. Libre Radic Res. 1999; 31: 273-300.
86. Sheehan D, Meade G., Foley VM, Dowd CA. Estructura, función y evolución de las transferencias de glutatión: implicaciones para la clasificación de miembros nomamánicos de una antigua superfamilia enzimática. Biochem J. 2001; 360: 1 - 16.
87. Cho SG, Lee YH, Parque HS, Ryoo K, Kang KW, et al. La glutatión S-transferasa Mu modula las señales activadas por estrés suprimiendo la quinasa 1 reguladora de la señal de apoptosis. J Biol Chem. 2001; 276: 12749-12755.
88. Dorion S, Lambert H, Landry J. La activación de la vía de señalización p38 por choque térmico implica la disociación de la glutatión S-transferasa Mu de Ask1. J Biol Chem. 2002; 277: 30792-30797.
89. Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, y col. Regulación de la señalización JNK por GSTp. EMBO J. 1999; 18: 1321-1334.
90. Manevich Y, Feinstein SI, Fisher AB. La activación de la enzima antioxidante 1-CYS peroxiredoxina requiere glutationilación mediada por heterodimerización con pGST. Proc Natl Acad Sci US A. 2004; 101: 3780 - 3785.
91. Bunker VW. Radicales libres, antioxidantes y envejecimiento. Med Lab Sci. 1992; 49: 299-312.
92. Mezzetti A, Lapenna D, F Romano, Costantini F, Pierdomenico SD, et al. Estrés oxidativo sistémico y su relación con la edad y la enfermedad. J Am Geriatr Soc. 1996; 44: 823-827.
93. Blanco E, Shannon JS, Patterson RE. Relación entre vitamina y
el uso de suplementos de calcio y el cáncer de colon. Cáncer Epidemiol Biomarcadores Prev. 1997; 6: 769-774.
94. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Nuevos mecanismos de compuestos antioxidantes naturales en sistemas biológicos: implicación del glutatión y enzimas relacionadas con el glutatión. J Nutr Biochem. 2005; 16: 577-586.
95. Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Cambios en el estado de glutatión cardiaco tras isquemia y reperfusión. Experientia. 1985; 41: 42-43.
96. El-Agamey A, Lowe GM, McGarvey DJ, Mortensen A, Phillip DM, Truscott TG. Química radical de los carotenoides y propiedades antioxidantes / pro-oxidantes. Arch Biochem Biophys. 2004; 430: 37-48.
97. Rice-Evans CA, Sampson J., Bramley PM, Holloway DE. ¿Por qué esperamos que los carotenoides sean antioxidantes in vivo? Libre Radic Res. 1997; 26: 381-398.
98. Niles RM. Vías de señalización en la quimioprevención retinoide y el tratamiento del cáncer. Mutat Res. 2004; 555: 81-96.
99. Donato LJ, Noy N. Supresión del crecimiento del carcinoma mamario por el ácido retinoico: los genes proapoptóticos son dianas para el receptor del ácido retinoico y la señalización de la proteína II de unión al ácido retinoico celular. Cancer Res. 2005; 65: 8193-8199.
100. Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, et al. Bcl-2 es un regulador clave para la muerte de la célula apoptótica inducida por el ácido retinoico en el neuroblastoma. Oncogene. 2006; 25: 5046-5055.
101. Dalton TP, Shertzer HG, Puga A. Regulación de la expresión génica por oxígeno reactivo. Ann Rev Pharmacol Toxicol. 1999; 39: 67-101.
102. Scandalios JG. Respuestas genómicas al estrés oxidativo. En: Meyers RA, ed. Enciclopedia de Biología Molecular de Células y Medicina Molecular. Vol 5. 2nd ed. Weinheim, Alemania: Wiley - VCH; 2004: 489-512.
103. Ghosh R, Mitchell DL. Home Idiomas Ingresar a Epistemonikos Búsqueda avanzada Efecto del daño oxidativo del ADN en los elementos promotores en el factor de transcripción vinculante. Nucleic Acids Res. 1999; 27: 3213-3218.
104. Marietta C, Gulam H, Brooks PJ. Una sola 8, 50-ciclo-20-desoxiadenosina lesión en una caja TATA impide la unión de la TATA vinculante proteína y reduce fuertemente la transcripción in vivo. Reparación del ADN (Amst). 2002; 1: 967-975.
105. Jackson AL, Chen R, Loeb LA. Inducción de inestabilidad de microsatélites
por daño oxidativo del ADN. Proc Natl Acad Sci US A. 1998; 95: 12468 - 12473.
106. Caldecott KW. Interacciones proteína-proteína durante la reparación de ruptura de una sola hebra de ADN de mamífero. Biochem Soc Trans. 2003; 31: 247-251.
107. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Daño oxidativo del ADN: mecanismos, mutación y enfermedad. FASEB J. 2003; 17: 1195-1214.
108. Jones PL, Wolffe AP. Relaciones entre la organización de la cromatina y la metilación del ADN en la determinación de la expresión génica. Semin Cancer Biol. 1999; 9: 339-347.
109. Girotti AW. Mecanismos de la peroxidación lipídica. J Free Radic Biol Med. 1985; 1: 87-95.
110. Siu GM, Draper HH. Metabolismo del malonaldehído in vivo e in vitro. Lípidos. 1982; 17: 349-355.
111. Esterbauer H, Koller E, Slee RG, Koster JF. Posible implicación del producto de peroxidación lipídica 4-hidroxinonenal en la formación de cromolípidos fluorescentes. Biochem J. 1986; 239: 405 - 409.
112. Hagihara M, Nishigaki I, Maseki M, Yagi K. Cambios dependientes de la edad en los niveles de peróxido lipídico en las fracciones de lipoproteínas del suero humano. J Gerontol. 1984; 39: 269-272.
113. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, et al. 4- Hidroxinonenal, un producto aldehídico de la peroxidación lipídica de la membrana, impide el transporte del glutamato y la función mitocondrial en los sinaptosomas. Neurociencia. 1997; 806: 85-96.
114. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activación de las vías de señalización del estrés por el producto final de la peroxidación lipídica. 4-hydroxy-2-nonenal es un potencial inductor de la producción de peróxido intracelular. J Biol Chem. 1999; 274: 2234-2242.
115. Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Activación del receptor de EGF por LDL oxidada. FASEB J. 1998; 12: 665-671.
116. 4-Hydroxy-2-nonenal mejora la producción de fibronectina por los fibroblastos de pulmón humano IMR-90 en parte a través de la activación de la quinasa regulada por señal extracelular unida al receptor del factor de crecimiento epidérmico p44 / 42 vía. Toxicol Appl Pharmacol. 2002; 184: 127-135.
117. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. Exhalado 8-isoprostano como biomarcador in vivo de estrés oxidativo pulmonar en pacientes con EPOC y fumadores saludables. Am J Respir Crit Care Med. 2000; 162: 1175-1177.
118. Morrison D, Rahman I, Lannan S, MacNee W. Permeabilidad epitelial, inflamación y estrés oxidante en los espacios de aire de los fumadores. Am J Respir Crit Care Med. 1999; 159: 473-479.
119. El aumento del contenido de sustancias reactivas al ácido tiobarbitúrico y peróxido de hidrógeno en el condensado expirado del aliento de pacientes con enfermedad pulmonar obstructiva crónica estable: ningún efecto significativo del tabaquismo. Respir Med. 1999; 93: 389-396.
120. Kelly FJ, Mudway ES. Oxidación de proteínas en la interfase aire-pulmón. Aminoácidos. 2003; 25: 375-396.
121. Dean RT, Roberts CR, Jessup W. Fragmentación de los polipéptidos extracelulares e intracelulares por los radicales libres. Prog Clin Biol Res. 1985; 180: 341-350.
122. Keck RG. El uso de hidroperóxido de t-butilo como una sonda para la oxidación de metionina en proteínas. Anal Biochem. 1996; 236: 56-62.
123. Davies KJ. Daño a las proteínas y degradación por radicales de oxígeno. I. Aspectos generales. J Biol Chem. 1987; 262: 9895-9901.
124. Stadtman ER. Oxidación catalizada por iones metálicos de las proteínas: mecanismo bioquímico y consecuencias biológicas. Gratis Radic Biol Med.
1990; 9: 315-325.
125. Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inactivación de enzimas metabólicas clave mediante reacciones de oxidación de función mixta: posible implicación en la renovación y el envejecimiento de las proteínas. Proc Natl Acad Sci US A. 1983; 80: 1521 - 1525.
126. Stadtman ER, Moskovitz J, Levine RL. Oxidación de residuos de metionina de proteínas: consecuencias biológicas. Señal Redox De Antioxid. 2003; 5: 577-582.
127. Stadtman ER, Levine RL. Oxidación por radicales libres de aminoácidos libres y residuos de aminoácidos en proteínas. Aminoácidos. 2003; 25: 207-218.
128. Stadtman ER. Oxidación de proteínas en el envejecimiento y enfermedades relacionadas con la edad. Ann NY Acad Sci. 2001; 928: 22-38.
129. Shacter E. Cuantificación y significado de la oxidación de proteínas en muestras biológicas. Medicamento Metab Rev. 2000; 32: 307-326.
130. Poli G, Leonarduzzi G, F Biasi, Chiarpotto E. Estrés oxidativo y señalización celular. Curr Med Chem. 2004; 11: 1163-1182.
131. Neufeld G, Cohen T., Gengrinovitch S, Poltorak Z. Factor de crecimiento endotelial vascular (VEGF) y sus receptores. FASEB J. 1999; 13: 9-22.
132. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, et al. Regulación de la generación de especies de oxígeno reactivo en fibroblastos por Rac1. Biochem J. 1996; 318: 379 - 382.
133. Sun T, Oberley LW. Regulación redox de los activadores transcripcionales. Gratis Radic Biol Med. 1996; 21: 335-348.
134. Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martínez-Galesteo E, Barcena JA, Lamas S. Regulación de Redox de unión de ADN c-Jun por S-glutatiolación reversible. FASEB J. 1999; 13: 1481-1490.
135. Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen Heininger
YM. Detección in situ de proteínas S-glutathionylated después de la derivatización de cisteína catalizada por glutaredoxina-1. Biochim Biophys Acta. 2006; 1760: 380-387.
136. Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulación de la glutaredoxina-1
expresión en un modelo de ratón de la enfermedad alérgica de las vías respiratorias. Am J Respir Cell Mol Biol. 2007; 36: 147-151.
137. Filomeni G, Rotilio G, Ciriolo MR. Señalización celular y el sistema redox glutatión. Biochem Pharmacol. 2002; 64: 1057-1064.
138. Pande V, Ramos MJ. Reconocimiento molecular de 15-deoxydelta (12,14) prostaglandina J (2) por el factor nuclear-kappa B y otras proteínas celulares. Bioorg Med Chem Lett. 2005; 15: 4057-4063.
139. Perkins ND. Integración de las vías de señalización celular con función NF-kappaB y IKK. Nat Rev Mol Cell Biol. 2007; 8: 49-62.
140. Gilmore TD. Introducción a NF-kappaB: jugadores, vías, perspectivas. Oncogene. 2006; 25: 6680-6684.
141. Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distintas funciones de la tiorredoxina en el citoplasma y en el núcleo. Un mecanismo de dos pasos de regulación redox del factor de transcripción NF-kappaB. J Biol Chem. 1999; 274: 27891-27897.
142. Ward PA. Papel del complemento, quimiocinas y citocinas reguladoras en la lesión pulmonar aguda. Ann NY Acad Sci. 1996; 796: 104-112.
143. Akira S, Kishimoto A. NF-IL6 y NF-kB en la regulación de genes de citoquinas. Adv Immunol. 1997; 65: 1-46.
144. Meyer M, Schreck R, Baeuerle PA. H2O2 y antioxidantes tienen efectos opuestos sobre la activación de NF-kappa B y AP-1 en células intactas: AP-1 como factor secundario antioxidante-sensible. EMBO J. 1993; 12: 2005-2015.
145. Abate C, Patel L, Rausher FJ, Curran T. Regulación de Redox de la actividad de unión de ADN fos y jun in vitro. Ciencia. 1990; 249: 1157-1161.
146. Galter D, Mihm S, Droge W. Efectos distintivos del disulfuro de glutatión sobre los factores de transcripción nuclear kB y la proteína activadora 1. Eur J Biochem. 1994; 221: 639-648.
147. Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. La actividad transcripcional de AP-1 está regulada por una asociación directa entre la tiorredoxina y Ref-1. Proc Natl Acad Sci US A. 1997; 94: 3633 - 3638.
Publicar descargo de responsabilidad *
Alcance de la práctica profesional *
La información aquí contenida en "El Paso, TX Estrés Oxidativo y Defensa Antioxidante" no pretende reemplazar una relación personal con un profesional de la salud calificado o un médico con licencia y no es un consejo médico. Lo alentamos a que tome decisiones de atención médica basadas en su investigación y asociación con un profesional de la salud calificado.
Información del blog y debates sobre el alcance
Nuestro alcance informativo se limita a la quiropráctica, musculoesquelética, medicina física, bienestar, contribuyendo etiológico alteraciones viscerosomáticas dentro de las presentaciones clínicas, la dinámica clínica del reflejo somatovisceral asociado, los complejos de subluxación, los problemas de salud delicados y/o los artículos, temas y debates de medicina funcional.
Brindamos y presentamos colaboración clínica con especialistas de diversas disciplinas. Cada especialista se rige por su ámbito de práctica profesional y su jurisdicción de licencia. Utilizamos protocolos funcionales de salud y bienestar para tratar y apoyar la atención de lesiones o trastornos del sistema musculoesquelético.
Nuestros videos, publicaciones, temas, asuntos e ideas cubren cuestiones clínicas, problemas y temas que se relacionan y respaldan directa o indirectamente nuestro ámbito de práctica clínica.*
Nuestra oficina ha intentado razonablemente proporcionar citas de apoyo y ha identificado el estudio o los estudios de investigación relevantes que respaldan nuestras publicaciones. Proporcionamos copias de los estudios de investigación de respaldo disponibles para las juntas reguladoras y el público a pedido.
Entendemos que cubrimos asuntos que requieren una explicación adicional de cómo puede ayudar en un plan de atención o protocolo de tratamiento en particular; por lo tanto, para discutir más a fondo el tema anterior, no dude en preguntar Dr. Alex Jiménez, DC, o póngase en contacto con nosotros en 915-850-0900.
Estamos aquí para ayudarlo a usted y a su familia.
Bendiciones
El Dr. Alex Jimenez corriente continua MSACP, enfermero*, CCCT, IFMCP*, CIFM*, ATN*
email: coach@elpasomedicinafuncional.com
Licenciado como Doctor en Quiropráctica (DC) en Texas & New Mexico*
Número de licencia de Texas DC TX5807, Nuevo México DC Número de licencia NM-DC2182
Licenciada como Enfermera Registrada (RN*) en Florida
Licencia de Florida N.° de licencia de RN RN9617241 (Control No. 3558029)
Estado compacto: Licencia multiestatal: Autorizado para ejercer en 40 Estados*
Matriculado actualmente: ICHS: MSN* FNP (Programa de enfermera practicante familiar)
Dr. Alex Jiménez DC, MSACP, RN* CIFM*, IFMCP*, ATN*, CCST
Mi tarjeta de presentación digital
